
Enabling Fast and Appropriate Drug-Product Supply for Phase I Clinical Trials

The competitive landscape in early drug development is especially prominent in meeting unmet patient needs. In areas where there is a more urgent need to develop orphan drugs, such as specific oncology and central nervous system indications, getting ahead means showing an efficacy signal as early in clinical development as possible. And while new and emerging biotechs are experts on the science of their disease focus, they often do not have the capabilities and industry experience to convert the concept into a high-quality drug substance and an appropriate and consistent dosage form.

In addition, the target patient population is often more difficult to find around the globe, thereby intensifying the need to understand regional regulatory requirements in multiple countries. It is important the drug product you make arrives at the clinical sites at the right time and intact (e.g., protected against high-shipping temperatures) to support patient dosing requirements. Therefore, while the reward of a successful start-up can be substantial, these ventures come with considerable risks and uncertainties. Looking at the historical trend in R&D costs per approved compound (see Figure 1), there has been a relentlessly upward trend in the cost to go from discovery to an approved product.
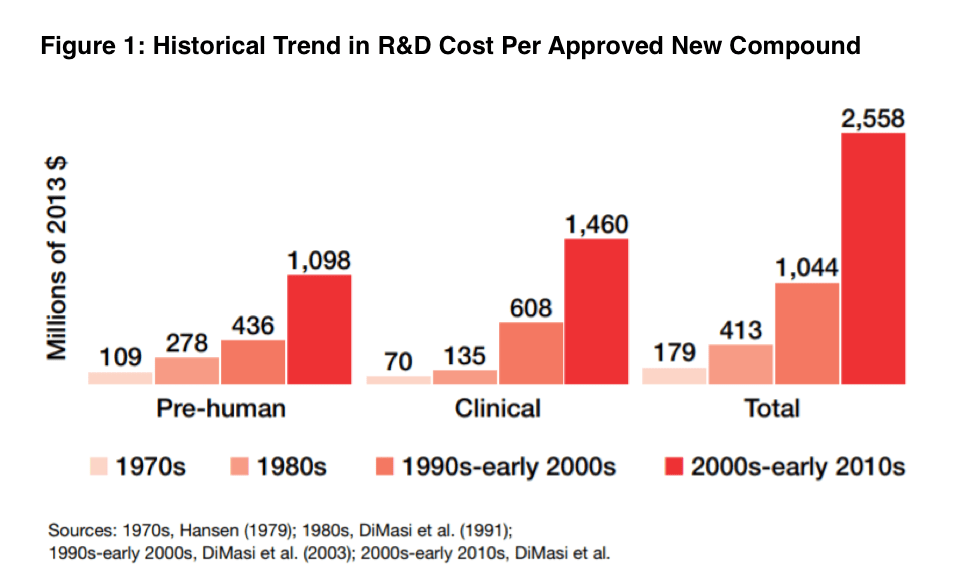
This challenge is exacerbated by the fact that only about 14% of all drugs in clinical trials eventually win approval from the FDA. For small drug companies pursuing a small molecule, this means they must beat their competitors to market with limited capital and a very small margin for error. Making it to Phase II, where dose response and efficacy signals of a drug candidate can emerge, marks a milestone that gives venture capitalists investing in the program more confidence that they will see a return on their investment. Doing so, though, requires early mitigation of product development risks that can help avoid clinical-trial failures later. This calls for a wide range of expertise, including extensive knowledge in chemistry, manufacturing, and controls (CMC); regulatory compliance; and clinical-trial management—resources that are often limited or not available in start-up pharma companies. Outsourcing is often the solution to fill these gaps, but it is important to work with a CDMO that incorporates cross-functional communication. For example, in Phase I, the focus is on identifying safety and tolerability at various dose levels, which will then need to be evaluated for potential side effects. However, a CDMO should not simply convert powder into an arbitrary dosage form without knowing the target patient population and any nuances associated with the therapeutic indication. This involves open discussion with clinical operations in conjunction with CMC development requirements. Questions worth exploring may include weight dosing and titration, home administration or site needs, countries targeted to ensure qualified person support in advance, short- and long-term stability, and temperature requirements.
Therefore, pharmaceutical scientists should be interacting with those in clinical-trial supply to break down silos that could lead to inefficiencies and a slower path to clinic. Recognizing the importance of aligning formulation, drug product manufacturing, and clinical-trial-management activities, Thermo Fisher Scientific developed the Quick to Clinic™ program. This service is designed to reduce the timeline to deliver Phase I supplies using a wide range of resources so that new and emerging biotechs can meet the crucial milestones for their project without compromising quality.