
Addressing Challenges with Clinical In-Use Testing of Biotherapeutics
Ensuring patient safety in early and late-phase product development
By Léa Sorret, Principal Scientist & Group Lead in Formulation Development Drug Product Services, Lonza
Introduction
All clinical in-use testing of biotherapeutics aims to develop a stable and safe drug product (DP) and is a regulatory requirement under ICH Q1A guidelines [1] for filing a registration application during early- and late-stage/commercial development. Testing includes stability and compatibility studies of the materials and diluents used during the in-use period of the DP comprising of the preparation, storage, and simulated administration, which may vary depending on the route of administration and the type of molecule(s) evaluated.
This testing helps ensure patient safety by identifying potential risks due to exposure of not-previously assessed contact materials, diluents or holding times at differing temperatures. The studies must also demonstrate the stability of the DP after reconstitution and/or dilution, as well as within the proposed administration dosage ranges to ensure that patients receive an accurate dose of DP at an appropriate product quality. Data obtained from clinical in-use testing should show that the administration procedure of a DP is safe, viable and accurate during clinical and commercial use. This information can be used to support a registration application, such as an Investigational New Drug (IND), Investigational Medicinal Product Dossier (IMPD) or market authorization filing.
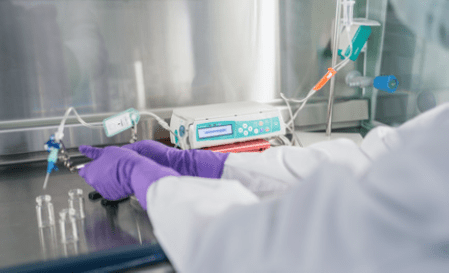
Figure 1
Simulated administration and sample collection in a laboratory.
In this article, we discuss the more demanding aspects surrounding clinical in-use testing of biotherapeutic DPs for parenteral delivery while reviewing potential strategies to address these issues. We include technical considerations a biopharma company must handle. Additionally, we illustrate how partnering with Lonza may help to address some of the challenges associated with clinical in-use testing workflows.
What are the Major Technical Challenges?
DP stability in commercial diluents
Biotherapeutic DP formulations are commonly optimized to ensure DP and drug substance (DS) stability to obtain an appropriate shelf life. However, for administration, the DP is often diluted using an infusion diluent to ensure each individual patient is correctly dosed, as typically a DP is supplied at one concentration which may not fit the required dosing of all the patient population(s).
Commonly used commercial diluents are saline or dextrose, which can compromise drug stability upon DP dilution, for example, due to high ionic strength or significant dilution of the formulation and its stabilizing excipients. As such, their compatibility should be assessed prior to use for administration. Using an incorrect diluent can result in loss of purity, protein aggregation/ particle formation or adsorption, which in turn can cause incorrect dosage or in the worst-case scenario may result in unwanted immune responses, leading to safety and efficacy issues [2].
Material compatibility
During in-use administration, the DP is introduced to many new contact materials using materials for dose preparation (including syringes, filters or filter-needles) and administration (for example intravenous [IV] bags, syringes, in-line filters, administration lines, connectors and catheters). These materials can have a critical impact on a DP’s quality, efficacy and safety. Some materials can cause a protein to adsorb to the administration system, which can lead to incorrect dosing and sub-optimal efficacy of the drug. The presence of leachables and extractables can also cause a DP to undergo protein aggregation, to form particles, or to degrade. Studies have shown that leachables from an IV bag containing a saline diluent for example can negatively affect the stability of a protein-based biotherapeutic [3].
Introduction of CSTDs (closed system transfer devices)
CSTDs are required to be used to protect patients and clinical or pharmacy personnel when they are handling or administering hazardous or highly potent drugs, such as antibody-drug conjugates (ADCs). CSTDs are being requested more often by clinical sites for, at-times, unnecessary use (with non-highly potent and/ or non-hazardous DPs), which is increasing the complexity of testing and likelihood of compatibility failure due to various liabilities discussed in this white paper.
CSTDs are usually composed of multiple components that can be used for drug preparation and administration. There are numerous FDA approved and unapproved CSTDs commercially available for parenteral use. To date, regulatory approval for CSTDs has been based solely on their ability to prevent “leakage” or provide a safety barrier. However, using CSTDs can lead to potential issues, which include contamination with extractable and leachable particles that can migrate into the DP and dosing solution, as well as drug adsorption and material incompatibility. In fact, a recent study of four different CSTDs [4] showed that the majority had leaks varying in size, and that some generated visible particles (caused by stopper coring) and sub-visible particles (due to silicone oil and particulate contamination), which could be introduced into the biotherapeutic DP and have the potential to cause safety and/ or dosing issues.
Due to their complex geometry and proprietary configuration, material-based compatibility testing is not possible, and each system must be tested independently, adding further constraints during simulated in-use and administration testing.

Low dose delivery accuracy and related analytical challenges
Often, dose escalation studies are performed in First-In-Human (FIH) and early clinical trials. In addition, highly potent biotherapeutics are often effective at extremely low doses. However, this poses challenges when trying to perform clinical in-use testing to support such scenarios. The first issue is that protein-based therapies tend to adsorb to material interfaces. Given the relatively large contact surface area, as well as the various contact materials dosing solutions are exposed to from dose preparation to administration, this can lead to potential product loss and subsequent under-dosing of patients. An adsorption problem can be further exacerbated when the DP is extensively diluted to prepare the final dosing solution, especially when the concentration of surface-protective excipients (such as surfactants) that are typically present in a DP formulation is as a result, diluted to concentrations below effective levels. An additional issue around dose accuracy is that the available analytical methods may face limitations to accurately quantify the lowest effective dosing solution, and this makes it difficult to precisely predict potency, as well as purity levels.
Furthermore, low concentration dosages can become particularly challenging when complex additional systems, such as CSTDs, are used during drug preparation and administration [4]. Inaccurate dosing when using these devices has been reported due to incomplete withdrawal of the product from the primary DP container or incomplete transfer of the dose into the infusion container due to increased hold-up volumes or specific device designs.
Generation of particles
Visible and sub-visible particles may represent safety and immunogenicity risks to patients and are tightly controlled and regulated during in-use administration [5,6,7]. These particles can be classified typically into two categories. Firstly, extrinsic particles can be introduced when handling the DP during dose preparation and administration (see Fig. 2). The second type are intrinsic (proteinaceous) particles that can be generated due to incompatibility during in-use and simulated administration procedures (see Fig. 2). Both contributions to particle generation need to be carefully assessed to ensure compliance with regulations.
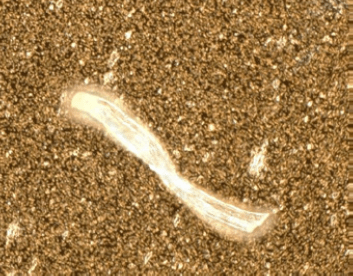
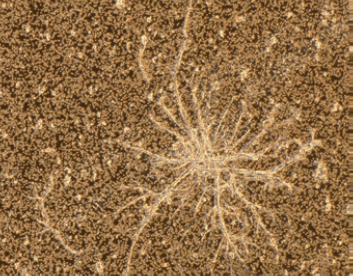

Figure 2
Microscopic image of (top) cellulose fiber, (center) proteinaceous aggregate, (bottom) polyethylene particle that can be generated during dose preparation and administration.
Clinical and regulatory challenges
In addition to technical challenges faced during clinical in-use testing, there are also clinical and regulatory hurdles to overcome. The complexity of the clinical set-up is one aspect that is difficult to control. An experienced Contract Development and Manufacturing Organization (CDMO) can make recommendations on suitable material and procedures for drug handling, dose preparation, and drug administration, typically summarized in a pharmacy manual. Additionally, local resources, training and regulations may mean that clinical in-use testing steps in standard pharmacy manuals may need to be adapted to fit local customer needs representing on the other hand potentially significant challenges to global clinical trials. A close interaction between clinical and technical development functions is required to ensure that the simulated in-use/ administration testing is closely aligned with the actual clinical procedures.
Furthermore, no universal global standards are available to support clinical in-use testing which means that technical data required to support in-use administration often needs to be tailored to the region of interest, adding complexity and supplementary effort from the drug sponsor.
To discover how to overcome these technical, clinical and regulatory challenges, read the rest of the technical article here.
References
[1] ICH Q1A (R2) Stability testing of new drug substances and drug products – Scientific guideline 2003 https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf [2] Luo S, McSweeney KM, Wang T, Bacot SM, Feldman GM, Zhang B. Defining the right diluent for intravenous infusion of therapeutic antibodies. MAbs. 2020; 12 (1):1685814. https://doi.org/10.1080/19420862.2019.1685814 [3] Chang JY, Xiao NJ, Zhu M, Zhang J, Hoff E, Russell SJ, Katta V, Shire SJ. Leachables from saline-containing IV bags can alter therapeutic protein properties. Pharm Res. 2010; 27(11):2402-13. https://doi.org/10.1007/s11095-010-0193-8 [4] Besheer A, Mahler HC, Matter-Schwald A, Barrenechea SM, Vogt M, Chalus P, Heymes P, Pillow T, Kirste A, Favrod P, Joerg S, Mathaes R. Evaluation of Different Quality-Relevant Aspects of Closed System Transfer Devices (CSTDs). Pharm Res. 2020; 109 (1):761-768. [5] United States Pharmacopeia (2023). General Chapter, {787} Subvisible Particulate Matter in Therapeutic Protein Injections. USP-NF. Rockville, MD: United States Pharmacopeia https://doi.org/10.31003/USPNF_M6497_02_01 [6] United States Pharmacopeia (2019). General Chapter, {788} Particulate Matter in Injections. Rockville, MD: US Pharmacopeia. https://www.uspnf.com/sites/default/files/usp_pdf/EN/USPNF/revisionGeneralChapter788.pdfhttps://doi.org/10.1007/s11095-020-02784-1 [7] United States Pharmacopeia (2022). General Chapter, {790}Visible Particulates in Injections. USP-NF. Rockville, MD: United States Pharmacopeia. https://doi.org/10.31003/USPNF_M7197_01_01